First Presentation of a MED12 Mutation Causing Capillary Malformation and Hemihyperplasia
AUTHORS:
Jacob B Diamond, MS; Thao Vu, MD; Kendall R Steadmon, MD
University of Florida Shands Children’s Hospital, Gainesville, FL
CASE REPORT | PUBLISHED SPRING 2022 | Volume 42, Issue 2
DOWNLOAD PDF
Abstract
A newborn born to a healthy mother with no complications was found to have extensive unilateral capillary malformations resembling port wine stains as well as right hemihyperplasia of the upper and lower extremities. While several genetic syndromes are associated with one of these findings, both occurring together is a rare, potentially novel presentation. The patient’s DNA was examined for mutations correlated with capillary malformations and hemihyperplasia, and while no such changes were found, a variant of unknown significance in a gene known to cause other genetic disorders was found. This may be the first description of a new MED12-related disorder.
Introduction
Vascular lesions in the newborn population are not uncommon, frequently regress on their own, and have no associated pathological features, although some are specifically correlated with syndromic disorders. These anomalies encompass everything from vascular tumors, simple and combined malformations and major vessel anomalies to syndromic-specific malformations. Infantile hemangioma (IH), a type of benign vascular tumor, is recognized as the most common newborn vascular lesion with an estimated incidence of 5%, while port wine stain (PWS), associated most often with Sturge Weber syndrome (SWS), is found in only three to five children per 1000 live births1,2. PWS, unlike IH, often does not regress in childhood. Contrastingly, the lesions’ color tends to darken from red to purple and even become raised and hardened after several decades. This is due to progressive dilatation of immature venule-like structures and subsequent formation of vascular nodules.
Hemihyperplasia (replacing the former term hemihypertrophy) is defined as one or several isolated areas of overgrowth of the body and represents another finding most commonly first reported at birth, with several established syndromic associations, most commonly Beckwith-Wiedemann syndrome (BWS). Although the prevalence of hemihyperplasia has been approximated at 1 in 86,000, the rate of isolated, non-syndromic findings consistent with hemihyperplasia has been more difficult to elucidate3. This case details a patient born with isolated findings of PWS and hemihyperplasia, as well as a genetic mutation of unknown significance, perhaps the first such composition of findings described in the literature.
Patient Presentation
A Caucasian female infant was born via uncomplicated spontaneous vaginal delivery at 37 weeks gestation to a 25-year-old gravida 2 para 1 mother, following an uncomplicated pregnancy. Delayed cord clamping and skin-to-skin contact were performed as part of routine care. The infant was determined to be large-for-gestational age (38 weeks estimated gestational age by Ballard score), but otherwise a healthy baby girl. Apgar scores were 9 and 9 at one and five minutes, respectively. At time of delivery, confluent, blanchable regions of erythema and purpura consistent with port-wine stains (PWS) were noted across the neonate’s trunk and right upper and lower extremities. The cutaneous defect was not identified anywhere on the patient’s face. The remainder of the newborn physical examination was normal. Routine newborn laboratory studies, including cord and venous blood gas, transcutaneous bilirubin, and Coombs antibody test were within normal limits. There was mild hypoglycemia that corrected upon administration of oral glucose gel and the neonate was started on formula feeds per mother’s request.
During her stay in the hospital, dermatology was consulted and described large segmental capillary malformations, consistent with PWS, of the right upper extremity (Figure 1), right lower extremity (Figure 2), and the lumbosacral area (Figure 3). Interestingly, the dermatology consult team also noted subtle overgrowth with moderate fullness of the soft tissues in the right upper extremity, specifically the right shoulder and axilla where the defect appeared. There was concern about several congenital disorders, but because the patient had no neurological deficits, was feeding and voiding properly, and in no acute distress, no further evaluation was warranted. Follow-up with outpatient dermatology and a lumbosacral ultrasound were scheduled and the infant was discharged home with her mother less than 48 hours after birth.
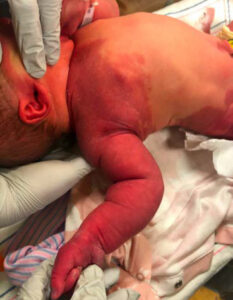 Figure 1 |
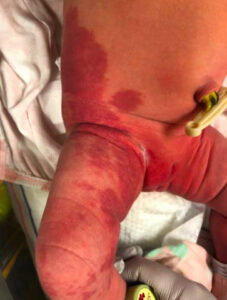 Figure 2 |
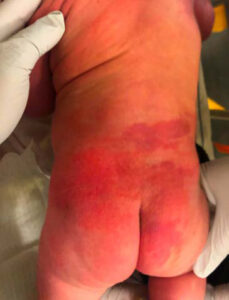 Figure 3 |
At 4 weeks of life the infant was meeting developmental milestones, continuing to feed and void regularly, and was progressing properly on length, weight, and head circumference growth percentiles. A spinal ultrasound showed no evidence of spinal cord tethering and was normal. Her physical examination findings were largely unchanged from birth with no changes in the growth or involvement of the PWS and no development of neurological deficits. The patient did show a moderate increase in circumference of the right proximal upper extremity compared to the left side. The patient was referred to a genetic counselor for further evaluation of the potential significance of the capillary malformation and unilateral arm overgrowth.
The genetic team first evaluated the patient at three months of age and recommended a buccal swab to be sent for pathologic gene abnormalities related to overgrowth as well as hemangiomas, including the macrocephaly-associated PIK3CA and GNAQ genes. Her DNA was negative for these two mutations; however the panel found a variant of unknown significance (VUS) in MED12, a gene that encodes a protein that regulates gene activity. Abnormalities in this gene are associated with several congenital disorders, none of which have findings consistent with this patient’s extensive capillary malformations and hemihypertrophy. As the patient was continuing to meet milestones appropriately at six months of age, the genetics team opted to delay further diagnostic testing, including skin biopsy, until her first birthday, or in the event of new or worsening symptoms.
Discussion
On initial evaluation, the genetics team was concerned primarily for macrocephaly-capillary malformation (M-CM) syndrome, caused by several PIK3CA gain of function mutations, which lead to superfluous activation of a cellular proliferation pathway4. Further investigation of neurocutaneous or congenital overgrowth disorders like SWS or BWS was not performed mainly due to the lack of other syndromic findings which are typically associated with these severe conditions. Diagnosis of M-CM syndrome depends on clinical findings of two major criteria (macrocephaly and capillary malformation) as well as one of several potential minor criteria, one of which is asymmetry or overgrowth4. While definitions in the literature vary slightly, the American Academy of Pediatrics denotes a head circumference >97th percentile as macrocephaly, and though the patient discussed here was nearing the upper percentiles (96th percentile at birth), she never met the clinical diagnostic criteria and by 3 months of age was within 1 standard deviation of normal at the 73rd percentile. Despite the lack of true macrocephaly, her DNA was tested for mutations in PIK3CA as well as GNAQ (most commonly associated with SWS). While these did not result in significant findings, a variant of unknown significance (VUS) was discovered in her mediator complex subunit 12 (MED12) gene. The initial impression that the patient’s unique presentation may represent a mosaic form of M-CM syndrome was likely refuted by the results of the buccal swab, as buccal epithelial and white blood cell samples have proven to be more sensitive when probing for mosaic patterns of mutation than their serum leukocytic counterparts5.
MED12 encodes a subunit of a multiprotein mediator complex which regulates the activity of transcription factors. Although its specific role in the transcription mediator protein multiplex is currently being studied, evidence points to its importance in cell growth, migration, and differentiation, specifically in developing neurons6. The exact VUS identified in the above patient was an A to G transition at base 6218 in the coding region, causing amino acid 2073 to change from glutamine to arginine. Specific mutations in the MED12 coding region have been linked to uterine leiomyomas as well as several genetic disorders including FG syndrome, Luhan syndrome, and a specific subtype of Ohdo syndrome7. These three syndromes are inherited in an X-linked recessive manner and share intellectual disability as a common finding, while each is notable for distinct physical features. Interestingly, limb size abnormalities, including hemihyperplasia, and capillary malformations are absent from the list of expected physical exam findings in any of these three syndromic disorders. With that being said, all three are exceedingly rare conditions and not commonly cited in the literature. While FG syndrome and Ohdo syndrome were first described in the late 1980s, Luhan syndrome was designated later, in the early 2000s. The vast majority of data collected on MED12, its associated functions, and the consequences of potential coding errors has occurred since 2007. The patient described above, with a mutation in the coding region and distinct physical abnormalities may represent the first-described case of a genetic disorder. The significance of her presentation at birth as well as the linkage of the physical findings to a genetic cause, including MED12, will become more evident as she ages, whether or not other symptoms develop including intellectual disability. If so, more genetic testing will be required.
References
- Kilcline C, Frieden IJ. Infantile hemangiomas: How common are they? A systematic review of the medical literature. Pediatr Dermatol. 2008;25(2):168-173. doi:10.1111/j.1525-1470.2008.00626.x
- Nguyen V, Hochman M, Mihm MC, Nelson JS, Tan W. The pathogenesis of port wine stain and sturge weber syndrome: Complex interactions between genetic alterations and aberrant MAPK and PI3K activation. Int J Mol Sci. 2019;20(9). doi:10.3390/ijms20092243
- Mutafoglu K, Cecen E, Cakmakci H. Isolated hemihyperplasia in an infant: An overlooked sign for wilms tumor development. Iran J Pediatr. 2010;20(1):113-117. Accessed September 2, 2020. /pmc/articles/PMC3446009/?report=abstract
- Mirzaa GM, Rivière JB, Dobyns WB. Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet Part C Semin Med Genet. 2013;163(2):122-130. doi:10.1002/ajmg.c.31361
- Gajecka M. Unrevealed mosaicism in the next-generation sequencing era. Mol Genet Genomics. 2016;291(2):513-530. doi:10.1007/s00438-015-1130-7
- Wang H, Shen Q, Ye L hua, Ye J. MED12 mutations in human diseases. Protein Cell. 2013;4(9):643-646. doi:10.1007/s13238-013-3048-3
- Lyons MJ. MED12-Related Disorders. University of Washington, Seattle; 1993. Accessed September 8, 2020. http://www.ncbi.nlm.nih.gov/pubmed/20301719