Disorders of Primary Hemostasis: Updates on Management
AUTHORS:
Sherri Huang, MD, PhD1; Sukjoo Cho, MD1; Jade Walter, DO1; Juan Felipe Rico, MD2*
1Department of Pediatrics, University of South Florida Morsani College of Medicine, Tampa, FL
2Division of Hematology/Oncology, Department of Pediatrics, University of South Florida Morsani
*Corresponding author
REVIEW ARTICLE | PUBLISHED WINTER 2023 | Volume 43, Issue 1
DOWNLOAD PDF
Abstract
In this review, we summarize updates on management of disorders of primary homeostasis, including immune thrombocytopenic purpura (ITP) and von Willebrand disease (VWD). The role of platelet transfusions in ITP and the management of abnormal uterine bleeding in VWD are also discussed.
Introduction
Hemostasis, the physiologic process of stopping bleeding, requires brisk generation of platelet plugs at the site of vascular injury and subsequent thrombin and fibrin formation. Disorders of primary hemostasis occur due to abnormal platelet plug formation. To produce effective platelet plugs, three key components are needed: a) adequate functional platelets, b) von Willebrand factor (vWF), the molecular glue mediating adhesion of platelets to the injured vessel wall, and c) a normal blood vessel constricting in response to damage (Table 1).1
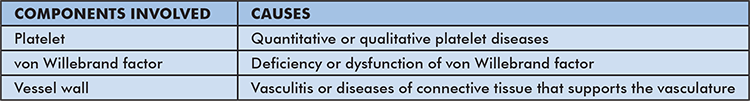
Table 1: Summarizes the common etiologies of disorders of primary hemostasis.
Thrombocytopenia can be caused by decreased production (e.g., bone marrow failure), increased destruction, or abnormal distribution (e.g., splenomegaly) of platelets. Increased destruction of platelets occurs via immune mechanisms, such as immune thrombocytopenic purpura (ITP), or non-immune mechanisms, such as microangiopathic diseases (e.g., thrombotic thrombocytopenic purpura, hemolytic uremic syndrome) and increased consumption (e.g., disseminated intravascular coagulation). Platelet dysfunction can occur via various mechanisms including abnormal adhesion or aggregation.
Von Willebrand disease (VWD) is the most common inherited bleeding disorder and is caused by deficient or defective vWF, resulting in abnormal platelet adhesion. Platelet dysfunction may also be related to acquired etiologies such as medications (e.g., aspirin or non-steroidal anti-inflammatory drugs) or azotemia. Blood vessel malfunction secondary to inflammation of the blood vessels (e.g., Henoch-Schonlein purpura) or connective tissue disorders (e.g., Marfan and Ehlers-Danlos syndromes) may also lead to bleeding.
Immune Thrombocytopenic Purpura
ITP is an immune-mediated condition that results in an isolated low platelet count of less than 100,000/mL with or without an underlying condition. In secondary ITP, underlying diseases such as human immunodeficiency virus or hepatitis C infection, systemic lupus erythematosus, or malignancy can trigger the type II hypersensitivity reaction leading to antibody-mediated destruction of platelets. In general, treatment indication depends on clinical stability of the patient, i.e., whether there is any moderate to severe bleeding. First-line treatment is corticosteroids. Intravenous immunoglobulin (IVIG) or anti-D immunoglobulin can be used in cases where corticosteroids are contraindicated or refractory. In recent years, thrombopoietin receptor agonists have also become established as standard treatment. Further recommendations on ITP management in pediatric patients are outlined below.
Updates in Guidelines
The American Society of Hematology (ASH) and University of Oklahoma Health Sciences Center in 2019 coordinated a panel of adult and pediatric hematologists along with two patient representatives to review and update the 2011 guidelines for management of ITP.2 Key features of pediatric ITP management in the 2019 compared to 2011 guidelines include:
- Regardless of platelet count, for children with newly diagnosed ITP who have no or mild bleeding, (i.e., skin manifestations only such as petechiae or purpura), outpatient over inpatient treatment is conditionally recommended. Exceptions include patients who have an uncertain diagnosis, social concerns, or potential poor follow-up; these patients may benefit from hospitalization. The provider should ensure that the patient has prompt follow-up with a hematologist within 24 to 72 hours of diagnosis.
- Both the 2011 and 2019 guidelines recommend that children with ITP who have minor or no bleeding should be observed versus having corticosteroids or IVIG administered (strong recommendation).
- For children with newly diagnosed ITP who have non-life-threatening mucosal bleeding (ie prolonged epistaxis or wet purpura) and/or diminished health-related quality of life, corticosteroids are preferred over IVIG or anti-D immunoglobulin (conditional recommendation).
- In children with ITP who have mucosal bleeding and/or decreased health-related quality of life, corticosteroids therapy should be 7 days or less (strong recommendation). Prednisone at 2-4 mg/kg per day with 120 mg daily maximum for 5-7 days is preferred over dexamethasone at 0.6 mg/kg per day with 40 mg per day maximum for 4 days (conditional recommendation).
- There is no preference for anti-D immunoglobulin versus IVIG.
- Thrombopoietin receptor agonist (TPO-RA), rituximab and splenectomy, are second-line therapies. In children who do not respond to first-line treatment (corticosteroids, IVIG or anti-D immunoglobulin), TPO-RAs are preferred over rituximab and splenectomy (conditional recommendation); rituximab is preferred over splenectomy (conditional recommendation).
Utility of platelet transfusion in ITP
In 1992, the British Paediatric Haematologic Group suggested restrictive platelet transfusion for the management of ITP (i.e., transfuse for life-threatening bleeding such as intracranial hemorrhage only).3 The group suggested a “watch and wait” for those patients with only bruising or petechiae rather than treating a number (i.e. platelets <10k-30k).3 The rationale behind this observational approach to children with ITP and no overt bleeding accounts for the low risk of bleeding without treatment; moreover, treatment does not change the natural history of the disease, and treatment can have harmful side effects. This approach was initially met with resistance, but over time has gained acceptance. In 1995, the proportion of children receiving medical treatment for ITP in the United Kingdom was 61% and fell to 16% by 2009.4 This approach was eventually adopted by ASH in 2011 and reiterated in the most recent ASH guidelines in 2019 as well as in the recommendations of a separate group, the “International consensus report on the investigation and management of primary immune thrombocytopenia.5-7” Switching to an observational approach for children with minimal bleeding symptoms did not influence bleeding rates or require treatment alteration in children with ITP in a single center study of over 300 children with ITP.8
Novel therapies for ITP
- Thrombopoietin receptor agonists
One of the most recent advances in ITP management is the class of medications called thrombopoietin receptor agonists (TPO-RAs) which include romiplostim, eltrombopag, avatrombopag, and lusutrombopag. TPO-RAs interact with the thrombopoietin (TPO) receptor on megakaryocytes to activate platelet production. The mechanism, efficacy and safety of TPO-RAs are
well-studied. In a mouse model, treatment with TPO resulted in increased platelet counts and reduced IgG anti-platelet antibodies, suggesting that TPO-RAs may also exert effects via immunomodulation.9 The efficacy and safety of self- versus healthcare professional administration of romiplostim was recently evaluated. Self-administration did not increase adverse effects and platelet responses were similar to those in patients administered romiplostim by healthcare professionals.10 A meta-analysis of the efficacy and safety of TPO-RAs in children with chronic ITP found that incidence of side effects was comparable to control groups and that the most common side effects were headaches, epistaxis, upper respiratory tract infection and nasopharyngitis.11
Immunomodulators
Other advances in the development of immunomodulators include fostamatinib, and efgartigimod, which is in phase 3 trials as of this writing. Fostamatinib is a tyrosine kinase inhibitor which was FDA-approved in 2018 for ITP. One study showed that 34% of patients who had failed TPO agents achieved response with fostamatinib.12 Efgartigimod, a human IgG1 antibody Fc-fragment that blocks neonatal Fc receptor and thus prevents recycling of IgG, has been shown to be well-tolerated in phase 2 trials. In addition, total IgG levels were reduced by 63.7% from baseline.13 Finally, Panzyga, a novel human IVIG 10% has been shown to be well-tolerated at high infusion rates with resultant rapid platelet increase in a clinical trial.14 These promising agents warrant further evaluation across age groups and in larger studies with TPO-RA refractory patients.
Prophylactic Platelet Transfusion
Given the critical role of platelets in primary hemostasis, prophylactic platelet transfusion is commonly performed to reduce the risk of bleeding in patients with thrombocytopenia. Most bleeding associated with low platelet counts is mild, although fatal bleeding involving critical sites such as lungs and brain may occur.15 The risk of spontaneous bleeding associated with thrombocytopenia is affected by multiple clinical factors including age (i.e., children and adolescents have overall higher bleeding risk compared to adults) 16, high fever, sepsis, disseminated intravascular coagulation, splenomegaly, vaso-occlusive disease, and history of chemotherapy or allogeneic hematopoietic stem cell transplantation transfusion.17 Patients with these clinical factors may require more aggressive prophylactic platelet transfusion (e.g., higher trigger threshold, larger and/or more frequent dose).
The threshold for prophylactic platelet transfusion has continued to evolve until recently. A threshold of 20,000/mL had been used for most patients until the 2000s. However, advances in the management of thrombocytopenic patients, particularly with those with infections, have resulted in lower thresholds.17 Several randomized controlled trials (RCTs) comparing the bleeding risk of a 10,000/mL vs 20,000/mL threshold demonstrated no difference.18-21 Such findings were confirmed in the Platelet Dosing (PLADO) study, a large multicenter RCT to establish prophylactic platelet transfusion dose; it also supported 10,000/mL as a prophylactic platelet transfusion threshold for stable hematology/oncology patients.15,22
Evidence supporting 10,000/mL as a trigger threshold in pediatric populations is less robust. The PLADO trial only included 198 children and adolescents out of 1,272 participants and the other RCTs listed above had limited pediatric involvement.22 Despite the paucity of pediatric data, both American Society of Clinical Oncology (ASCO) guidelines and International Collaboration for Transfusion Medicine Guidelines (ICTMG) endorse 10,000/mL as a reasonable trigger threshold for prophylactic platelet transfusion in children as well as adults.23,24
For neonates, the threshold for prophylactic platelet transfusion has been debated without consensus, leading to significant variation in transfusion policy among providers and centers. The multicenter RCT PlaNeT2 investigated the trigger for prophylactic platelet transfusion in this population.25 In this trial, 660 preterm infants with severe thrombocytopenia were randomly assigned to receive platelet transfusion when the platelet count was less than 50,000/mm3 (the high threshold group) vs. 25,000/mm3 (the low threshold group). The trial showed that major bleeding (e.g., intracranial hemorrhage) or death occurred more frequently in the high threshold group than in the counterpart, implying the benefits of the lower trigger threshold for prophylactic platelet transfusion.
Von Willebrand Disease
Von Willebrand disease (VWD) is the most common inherited bleeding disorder, affecting up to 1 percent of the population.26 It is a disorder of primary hemostasis caused by a defect or deficiency of the multimeric glycoprotein von Willebrand factor (vWF) leading to impaired platelet adhesion and aggregation to exposed collagen at sites of injury. vWF facilitates platelet binding via mediation of GPIIb/IIIa receptors, promoting platelet plug formation. vWF also acts as a chaperone for clotting factor VIII, preventing its breakdown.
VWD is classified into three types based on bleeding severity, inheritability, and a qualitative versus quantitative deficiency of vWF protein.27 Each type of VWD is caused by a wide spectrum of mutations affecting a variety of genetic loci. Type 1 is inherited in an autosomal dominant fashion and is a quantitative disorder characterized by a partial loss of vWF. It is the most common type of VWD, accounting for approximately 75% of patients. Type 2 is also autosomal dominant and is characterized by several qualitative abnormalities resulting in dysfunctional vWF. Type 2 is further categorized into four subtypes based on the genetic variants that cause each type. Type 3 is autosomal recessive and leads to the most severe disease with a nearly complete loss of vWF.
The presentation of VWD is characterized by excessive mucocutaneous bleeding such as heavy menstrual bleeding, epistaxis, easy bruising, prolonged bleeding from minor wounds, oral or gastrointestinal bleeding, or bleeding that may occur after dental procedures, childbirth, or surgery. Joint bleeding is generally only seen in severe cases.
Diagnosis
The management of VWD centers around the proper diagnosis, which can often be challenging due to wide variability in patient bleeding symptoms, variability in clinical practice, and lack of specific evidence to guide decision making. For this reason, the American Society of Hematology (ASH), the International Society on Thrombosis and Haemostasias (ISTH), the National Hemophilia Foundation (NHF), and the World Federation of Hemophilia (WFH) came together to develop evidence-based guidelines to ensure accurate diagnosis.28
As with any disorder, initial diagnosis begins with a strong history and physical. If there is clinical suspicion of a bleeding disorder but a low probability of VWD (often seen in the primary care setting), the recommendation is to first use a standardized bleeding assessment tool to determine if a patient needs specific blood testing.
For patients with intermediate risk (e.g., significant bleeding history such as heavy bleeding with surgical procedures, dental extractions and/or childbirth, frequent nosebleeds lasting more than ten minutes, and heavy and/or prolonged menstrual bleeding; abnormal initial labs) or high risk (e.g., affected first-degree relative) probability of VWD, the recommendation is to use specific blood testing versus a bleeding assessment tool. Blood testing should include a CBC, CMP, PT/PTT with or without fibrinogen, factor VIII level, total vWF antigen plasma levels (vWF:Ag) and tests of specific vWF platelet binding activity. VWD diagnostic testing should be performed when patients are at a baseline state of health as vWF is an acute-phase reactant.
Traditionally, the ristocetin co-factor assay (vWF:RCo) was the preferred test for von Willebrand activity. This assay applies the glycopeptide antibiotic ristocetin to induce vWF binding to glycoprotein Ib-IX-V on target platelets. However, ASH VWD 2021 guidelines recommend newer assays that more directly measure the platelet binding activity of vWF including vWF:GP1bM and vWF:GP1b.28 These tests are not yet widely available but likely will become increasingly available in the near future as they are currently recommended by the ASH guidelines.
ASH guidelines also specify the levels of vWF (i.e., vWF antigen; vWF:Ag) or platelet-dependent vWF activity in diagnosis. The full diagnostic algorithm of VWD is beyond the scope of this article and can be accessed in the 2021 ASH VWD guidelines. However, in general, vWF:Ag levels above 0.50 IU/ml rule out diagnosis of VWD. Patients with both levels 0.30 to 0.50 IU/ml and signs of bleeding, and patients with levels below 0.30 IU/ml should have their platelet-dependent vWF activity/vWF:Ag ratio measured, which may distinguish a possible type 1 VWD from a type 2 VWD diagnosis. In addition, to confirm a diagnosis of VWD type 1, ASH guidelines recommend using a vWF level below 0.30 IU/mL, regardless of bleeding, and a vWF level below 0.50 IU/mL in the presence of abnormal bleeding.
Management
Treatment of VWD focuses on stopping and preventing bleeding episodes. Management can include therapies that directly increase vWF levels, such as desmopressin and vWF concentrates, as well as adjuvant therapies, such as the antifibrinolytics, tranexamic acid or aminocaproic acid. There is significant variability in the treatment options offered to patients with VWD due in part to variability in clinical practice and the wide variability in an individual patient’s bleeding symptoms. As such, ASH, the International Society on Thrombosis and Haemostasis, the National Hemophilia Foundation, and the World Federation of Hemophilia formed a multidisciplinary panel to establish comprehensive evidence-based guidelines for the management of VWD.29 The guidelines provide evidence-based recommendations for patients and clinicians regarding prophylaxis for recurrent bleeding, desmopressin trials, use of antiplatelet agents and anticoagulants, target vWF and factor VIII activity levels for surgery or procedures, and management of heavy menstrual bleeding. Published in 2021, these guidelines provide an update to the Quick Reference Guide for Management of VWD published by ASH in 2012:
- In patients with VWD who have history of severe or recurrent bleeding, use long-term prophylaxis treatment rather than no prophylaxis (conditional recommendation).
- In patients with VWD who are eligible to receive desmopressin (primarily type 1 VWD, contraindicated in types 2B, and 3) and who have vWF levels <0.30 IU/mL, perform a desmopressin trial and treat based on the results, including the use of tranexamic acid or vWF concentrate (conditional recommendation).a. It is not recommended to treat eligible patients with desmopressin in the absence of a desmopressin trial.
- In patients with VWD and a history of cardiovascular disease, give the necessary antiplatelet or anticoagulant therapy versus no treatment (conditional recommendation).
- In patients with VWD undergoing major surgery, both Factor VIII and vWF activity levels should be >0.50 IU/mL for at least 3 days after surgery (conditional recommendation).a. Target both Factor VIII and vWF activity levels.b. This recommendation includes procedures where even a small amount of bleeding could critically affect organ function (e.g., central nervous system).c. These patients should not receive desmopressin as sole therapy.
- In patients with VWD undergoing minor surgery, increase vWF levels to ≥0.50 IU/mL with desmopressin or factor concentrate along with tranexamic acid and not just with desmopressin or factor concentrate alone (conditional recommendation).a. In patients with type 1 VWD with baseline activity levels >0.30 IU/mL and mild bleeding, consider using tranexamic acid alone versus increasing vWF levels to ≥0.50 IU/mL with desmopressin or factor concentrate if undergoing a minor surgery or procedure.b. Finally, throughout the course of treatment, it is imperative to reassess bleeding symptoms, bleeding risk, and the need for prophylaxis.
VWD and abnormal uterine bleeding
VWD affects males and females equally, however the presentation of abnormal uterine bleeding or heavy menstrual bleeding (HMB) in adolescence may alert providers to patients with potential VWD. Indeed, HMB is a common manifestation of VWD and is often the presenting symptom for patients with VWD in the primary care setting. Approximately 15-30% of women with HMB may have a bleeding disorder.30 HMB affects quality of life, can lead to absenteeism, and is responsible for 2/3 of hysterectomies performed during reproductive age. HMB is defined as a loss of greater than 80 mL of menstrual blood per cycle, which equates to having to change a tampon or pad more than once an hour, clots greater than one inch, soaking through pajamas/sheets at night, periods lasting seven days or longer, and a low ferritin (less than 10-15 based on age). If any one of these screening criteria are positive in a patient of reproductive age, ACOG recommends initiating a bleeding workup.31
ASH 2021 guidelines include recommendations for managing heavy menstrual bleeding in patients with VWD. Treatment options include combined hormonal contraception, levonorgestrel-releasing intrauterine system, and tranexamic acid and are recommended over desmopressin.29 In general, these patients should be regularly for iron deficiency and/or anemia. Additionally, women with known bleeding disorders and HMB should undergo a standard gynecological assessment to rule out pathologic causes (e.g., fibroids, polyps) of HMB.
Conclusions
In recent years, translational and clinical research advances in the field of primary homeostasis have resulted in ongoing updates to clinical care guidelines. ITP, VWD and heavy menstrual bleeding are the more common disorders of primary homeostasis that pediatricians are likely to encounter in practice. For the pediatrician, careful monitoring of guideline updates as well as adapting conditional recommendations based on individual patient assessment will lead to the best patient outcomes.
References
- Hoffman R, Benz EJ, Silberstein LE, et al. Hematology : basic principles and practice. Seventh edition. ed. Elsevier; 2018.
- Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. Dec 10 2019;3(23):3829-3866. doi:10.1182/bloodadvances.2019000966
- Eden OB, Lilleyman JS. Guidelines for management of idiopathic thrombocytopenic purpura. The British Paediatric Haematology Group. Arch Dis Child. Aug 1992;67(8):1056-8. doi:10.1136/adc.67.8.1056
- Grainger JD, Rees JL, Reeves M, Bolton-Maggs PH. Changing trends in the UK management of childhood ITP. Archives of disease in childhood. Jan 2012;97(1):8-11. doi:10.1136/adc.2010.184234
- Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. Apr 21 2011;117(16):4190-207. doi:10.1182/blood-2010-08-302984
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. Jan 14 2010;115(2):168-86. doi:10.1182/blood-2009-06-225565
- Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood advances. Nov 26 2019;3(22):3780-3817. doi:10.1182/bloodadvances.2019000812
- Schultz CL, Mitra N, Schapira MM, Lambert MP. Influence of the American Society of Hematology guidelines on the management of newly diagnosed childhood immune thrombocytopenia. JAMA pediatrics. Oct 2014;168(10):e142214. doi:10.1001/jamapediatrics.2014.2214
- Kapur R, Aslam R, Speck ER, Rebetz JM, Semple JW. Thrombopoietin receptor agonist (TPO-RA) treatment raises platelet counts and reduces anti-platelet antibody levels in mice with immune thrombocytopenia (ITP). Platelets. 2020;31(3):399-402. doi:10.1080/09537104.2019.1624709
- Kuter DJ, Newland A, Chong BH, et al. Romiplostim in adult patients with newly diagnosed or persistent immune thrombocytopenia (ITP) for up to 1 year and in those with chronic ITP for more than 1 year: a subgroup analysis of integrated data from completed romiplostim studies. Br J Haematol. May 2019;185(3):503-513. doi:10.1111/bjh.15803
- Guo JC, Zheng Y, Chen HT, et al. Efficacy and safety of thrombopoietin receptor agonists in children with chronic immune thrombocytopenia: a meta-analysis. Oncotarget. Jan 23 2018;9(6):7112-7125. doi:10.18632/oncotarget.23487
- Bussel JB, Arnold DM, Boxer MA, et al. Long-term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. Am J Hematol. May 2019;94(5):546-553. doi:10.1002/ajh.25444
- Newland AC, Sanchez-Gonzalez B, Rejto L, et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol. Feb 2020;95(2):178-187. doi:10.1002/ajh.25680
- Arbach O, Taumberger AB, Wietek S, Cervinek L, Salama A. Efficacy and safety of a new intravenous immunoglobulin (Panzyga((R)) ) in chronic immune thrombocytopenia. Transfus Med. Feb 2019;29(1):48-54. doi:10.1111/tme.12573
- Slichter SJ, Kaufman RM, Assmann SF, et al. Dose of prophylactic platelet transfusions and prevention of hemorrhage. N Engl J Med. Feb 18 2010;362(7):600-13. doi:10.1056/NEJMoa0904084
- Josephson CD, Granger S, Assmann SF, et al. Bleeding risks are higher in children versus adults given prophylactic platelet transfusions for treatment-induced hypoproliferative thrombocytopenia. Blood. Jul 26 2012;120(4):748-60. doi:10.1182/blood-2011-11-389569
- Triulzi DJ. How well do platelets prevent bleeding? Hematology Am Soc Hematol Educ Program. Dec 4 2020;2020(1):518-522. doi:10.1182/hematology.2020000136
- Rebulla P, Finazzi G, Marangoni F, et al. The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto. N Engl J Med. Dec 25 1997;337(26):1870-5. doi:10.1056/NEJM199712253372602
- Heckman KD, Weiner GJ, Davis CS, Strauss RG, Jones MP, Burns CP. Randomized study of prophylactic platelet transfusion threshold during induction therapy for adult acute leukemia: 10,000/microL versus 20,000/microL. J Clin Oncol. Mar 1997;15(3):1143-9. doi:10.1200/JCO.1997.15.3.1143
- Zumberg MS, del Rosario ML, Nejame CF, et al. A prospective randomized trial of prophylactic platelet transfusion and bleeding incidence in hematopoietic stem cell transplant recipients: 10,000/L versus 20,000/microL trigger. Biol Blood Marrow Transplant. 2002;8(10):569-76. doi:10.1053/bbmt.2002.v8.pm12434952
- Diedrich B, Remberger M, Shanwell A, Svahn BM, Ringden O. A prospective randomized trial of a prophylactic platelet transfusion trigger of 10 x 10(9) per L versus 30 x 10(9) per L in allogeneic hematopoietic progenitor cell transplant recipients. Transfusion. Jul 2005;45(7):1064-72. doi:10.1111/j.1537-2995.2005.04157.x
- Slichter SJ, Kaufman RM, Assmann SF, et al. Effects of Prophylactic Platelet (Plt) Dose on Transfusion (Tx) Outcomes (PLADO Trial). Blood. 2008;112(11):285-285. doi:10.1182/blood.V112.11.285.285
- Schiffer CA, Bohlke K, Delaney M, et al. Platelet Transfusion for Patients With Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. Jan 20 2018;36(3):283-299. doi:10.1200/JCO.2017.76.1734
- Nahirniak S, Slichter SJ, Tanael S, et al. Guidance on platelet transfusion for patients with hypoproliferative thrombocytopenia. Transfus Med Rev. Jan 2015;29(1):3-13. doi:10.1016/j.tmrv.2014.11.004
- Curley A, Stanworth SJ, Willoughby K, et al. Randomized Trial of Platelet-Transfusion Thresholds in Neonates. N Engl J Med. Jan 17 2019;380(3):242-251. doi:10.1056/NEJMoa1807320
- Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. Feb 1987;69(2):454-9.
- Veyradier A, Jenkins CS, Fressinaud E, Meyer D. Acquired von Willebrand syndrome: from pathophysiology to management. Thromb Haemost. Aug 2000;84(2):175-82.
- James PD, Connell NT, Ameer B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. Jan 12 2021;5(1):280-300. doi:10.1182/bloodadvances.2020003265
- Connell NT, Flood VH, Brignardello-Petersen R, et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. Jan 12 2021;5(1):301-325. doi:10.1182/bloodadvances.2020003264
- James PD. Women and bleeding disorders: diagnostic challenges. Hematology Am Soc Hematol Educ Program. Dec 4 2020;2020(1):547-552. doi:10.1182/hematology.2020000140
- ACOG committee opinion no. 557: Management of acute abnormal uterine bleeding in nonpregnant reproductive-aged women. Obstet Gynecol. Apr 2013;121(4):891-896. doi:10.1097/01.AOG.0000428646.67925.9a